October 24, 2019
October 24, 2019
Adverse drug reactions (ADRs) are a significant cause of deaths and emergency hospital visits. The good news is that monitoring and understanding ADRs can help minimize and even prevent such events from happening. This requires labs to place a greater emphasis on pharmacovigilance.
Pharmacovigilance (also PV or PhV) covers all the activities focused on ADRs. Its main aims are to:
Often, people relate pharmacovigilance with safety reporting. Whereas this is a major aspect, it is actually only one of the components of the pharmacovigilance process, which also involves generating data, risk management, and input from regulatory authorities.
The largest sources of such ADR data are physicians, pharmacists, other healthcare professionals, drug regulatory authorities, pharmaceutical companies, and patients themselves. Data from these sources comes from places like medical literature, articles, registries, clinical trials, epidemiological studies, and even social media (especially when from patients).
In all these cases, the main problems to overcome are:
These two factors mean that underreporting is common and that safety issues may not always be immediately apparent. This is particularly likely to be an issue if the adverse reaction only impacts a small percentage of patients, if symptoms are minor, or if it is unclear that symptoms are connected to the drug.
Once labs have the data, it’s time for the evaluation stage. This consists of receiving, triage, assessing, distributing, and archiving data.
Manufacturers of medications need to assess every case of adverse reaction for expectedness, seriousness, and causality. This may involve contacting the patient or healthcare provider for more information.
After an analysis, the manufacturer must decide on the urgency of the case and whether it is necessary to report the event to the appropriate regulatory authorities. (In the U.S., this is the FDA.) In the case the drug manufacturer decides that an ADR does not require a report to the regulatory authorities, the lab must still keep a report in its database.
When reporting an ADR, manufacturers must include at least the following:
Assessing and managing risks is a growing concern for pharmaceutical companies. The speed at which information now travels means that consumers are quickly aware of safety concerns with medications, even when an event takes place on the other side of the world.
Pharmacovigilance for labs prepares manufacturers for crises — both real and perceived. For instance, the FDA can issue a Form 483, which notifies the company’s management of objectionable conditions and can order a halt to the manufacturing of certain drugs. Pharmacovigilance lays out a plan for addressing concerns in a timely manner while avoiding unnecessary complications and undue caution. It also helps manufacturers prepare for the eventuality that a regulatory authority will suspend the sale of a drug or impose restrictions, such as only allowing sales under specific conditions.
Some facets of risk management include:
Regulatory authorities do not only play an active role for safety monitoring in drug development, they also conduct post-marketing surveillance. This uses distinct sources of information and infrastructure from the evaluation and approval phases. For instance, regulatory authorities consider the risk and harm assessments of initial evaluators, run active inquiries, and utilize clinical investigation tools.
In each country, the post-marketing surveillance process is slightly different. In the case of the U.S., the FDA appoints an ADR advisory committee made up of experts from various fields. This ensures that the drug is examined from a number of angles, including epidemiology, pediatrics, toxicology, clinical medicine, and pharmacology.
Confirming drug safety during clinical product development alone is always a challenge due to data limitations. If labs had unlimited time to get their drugs to market, it would be less of a problem. Unfortunately, manufacturers need to start selling as soon as possible to maximize profits.
However, by dedicating more resources to pharmacovigilance, labs can maintain patient safety and public trust for a few reasons.
It is unusual to be able to conduct a clinical trial on a sample of patients that is anywhere close to the same number as who will be taking the medication. This means that rare adverse reactions may not emerge until after clinical trials are over.
Similarly, most clinical trials are carried out on a particular population. Often, patients are from a certain country and fall into a narrow age group. Therefore, manufacturers lack information about how patients of different ethnicities and from special populations (such as pregnant, pediatric, and geriatric patients) will react to the drug.
Even the types of patients tested in clinical trials could react differently to the drug in a natural setting. This is because, in a clinical trial, patients must adhere to strict guidelines, which are impossible to enforce in the real world.
Furthermore, actual patients may be more complex than in the trial. They could be suffering from additional chronic conditions or taking other medications (or medicinal products, herbal products, or dietary supplements) that could interact with the drug. There is also the risk that patients could confuse the medication with another.
Finally, there is the fact that it is only possible to detect some adverse effects after prolonged exposure to the medication. Effects could only be apparent after several years — far longer than is feasible for any clinical trial.
Often, it is only possible to see that a drug has the potential for abuse or overdose after commercialization.
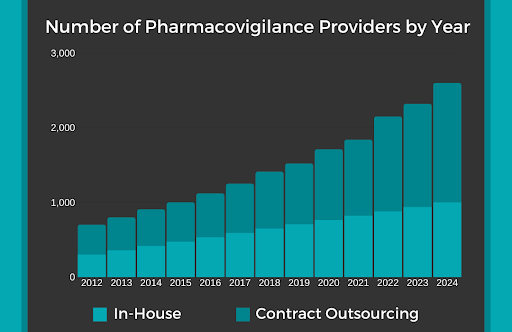
Most labs find that it is too challenging to handle all the aspects of pharmacovigilance in house. After all, even a minor error could have a huge impact on patients’ safety. As shown above, the amount of pharmacovigilance providers who are outsourced has increased exponentially throughout the years and is expected to continue until 2024. The solution is to receive pharmacovigilance services from a dedicated life sciences consultant.
ProPharma Group is a premier provider of pharmaceutical consulting. We ensure that drugs and medical devices meet safety standards to help labs maintain public confidence and trust.

November 16, 2023
Our "Meet the Expert" series introduces you to our team of experts around the world. This "behind the curtain" view will help you get to know who we are on a professional and personal level, and...

December 21, 2023
Our "Meet the Expert" series introduces you to our team of experts around the world. This "behind the curtain" view will help you get to know who we are on a professional and personal level, and...
September 3, 2021
Every marketing authorization holder (MAH) needs a pharmacovigilance system to guarantee the safety of its products. When setting up a pharmacovigilance system in Europe, the Qualified Person for...